|
Share this:
Read this article in pdf format
Biology of bile acids
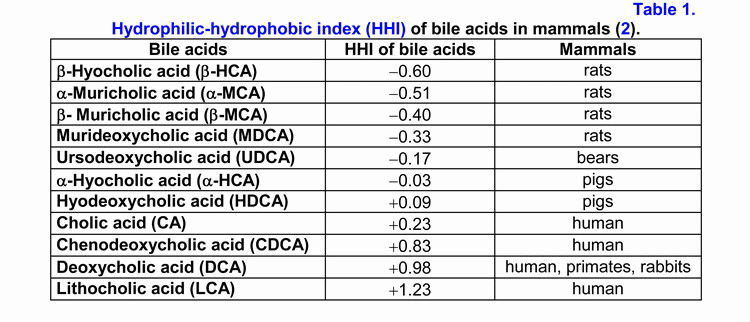
According to the hydrophilic-hydrophobic index, the bile acids are divided into hydrophilic and hydrophobic ones (table 1) (1-3).
If the hydrophobic index is less than that of cholic acid (CA), the bile acids are hydrophilic, if it is more than the hydrophobic index, they are hydrophobic (1-3). The primary bile acids are more hydrophilic than the secondary ones, but the taurine conjugates of the bile acids are more hydrophilic than the glycine ones (1-3). The hydrophilic bile acids have hepatoprotective properties [muricholic (MCA) > ursodeoxycholic (UDCA) > cholic (CA)] (4, 5). The hydrophobic bile acids are hepatotoxic [lithocholic (LCA) > deoxycholic (DCA) > chenodeoxycholic (CDCA) > CA] (1-7). Depending on the concentration, the hydrophobic bile acids cause cholestasis (LCA > DCA), necrosis (LCA > DCA) or apoptosis of hepatocytes (LCA > DCA > CDCA) (2-7).
Furthermore, DCA is cancerogenic (8). Experiments on animals showed that it causes cancer of the colon (9). The hydrophilic bile acids prevent the development of cholestasis or necrosis/apoptosis of hepatocytes (UDCA, MCA), as well as cancer of the colon (UDCA) (4-7, 9).
In serum up to 40% of bile acids are transported with HDL, up to 15% with LDL (10). The mechanism of binding of bile acids with lipoproteins depends on their hydrophobic index (CDCA > DCA > UDCA > CA > 7-epicholic acid) (10). In the liver, 60-80% of bile acids are uptake during one passage of portal blood (11). In earlier experiments on hamsters, it was demonstrated that the hepatic LDL uptake could influence the bile flow rate, the biliary secretion of bile acids and cholesterol (12, 13). The composition and concentration of bile acids participating in the enterohepatic circulation can modulate the LDL receptor activity and the receptor-dependent LDL uptake in the liver. More hydrophilic UDCA stimulates the receptor-dependent LDL uptake in the liver, but more hydrophobic CDCA decreases the LDL receptor activity (12, 13). It was also shown that the addition of hydrophobic CDCA to the hypercholesterolemic diet reduces the decrease of HDL concentration in serum, but the addition of hydrophilic UDCA causes the opposite effect (14, 15).
In hepatocytes, the bile acids may inhibit the activity of HMG-CoA reductase and cholesterol-7α-hydroxylase, depending on their concentration and hydrophobic index (DCA > CDCA > CA > UDCA) (2, 16-18). The hydrophilic bile acids stimulate the secretion of the hepatic bile (UDCA > CA), the hydrophobic ones lower it (LCA > DCA > CDCA) (19-21). UDCA and CDCA reduce the secretion of biliary cholesterol in the hepatic bile, but CA and DCA raise it (1, 19-21). In the gallbladder bile, the hydrophobic bile acids form mixed (bile acid-phospholipid-cholesterol) and simple (bile acid-cholesterol) micelles (DCA > CDCA > CA), but the hydrophilic bile acids form liquid crystalline lamellas (MCA > UDCA); that is, the lower the hydrophobic index of bile acids, the lower the ability to form micelles (22-25).
In the ileum, CA and CDCA raise the absorption of cholesterol, but UDCA and DCA reduce it (26-29). During the process of enterohepatic circulation, in the ileum and the colon, anaerobic bacteria promote 7α-dehydroxylation of the primary bile acids (hyocholic (HCA), MCA, CA, CDCA) and the formation of the secondary bile acids (hyodeoxycholic (HDCA), murideoxycholic (MDCA), DCA, LCA) (1, 2, 30, 31). The secondary bile acids are more hydrophobic than the primary ones (HDCA > HCA, MDCA > MCA, DCA > CA, LCA > CDCA) (1-3). The secondary bile acids are usually poorly absorbed in the ileum and the colon and are excreted with feces (1-3).
The mechanism of “lithogenic” bile formation
In patients with chronic calculous cholecystitis, there is a marked increase of the COX-2 expression. Previously, we had demonstrated the increased COX-2 expression in the gallbladder wall in the gallbladder specimens which were obtained from 21 patients with chronic calculous cholecystitis after cholecystectomy (32).
An increase of COX-2 expression in the gallbladder wall (n=21) was observed in 86% of the smooth muscle cells, in 81% of the epithelial cells, in 71% of the vascular smooth muscle cells, in 57% of the stromal cells and in 37% of the Rokitansky-Aschoff sinuses (32).
At mild degree of inflammation in the gallbladder wall (n=12), the COX-2 expression was increased in 83% of the epithelial cells, in 78% of the vascular smooth muscle cells, in 75% of the smooth muscle cells, in 33% of the stromal cells and in 17% of the Rokitansky-Aschoff sinuses.
At moderate and severe degree of inflammation in the gallbladder wall (n=9), the COX-2 expression was increased in 100% of the smooth muscle cells, in 89% of the vascular smooth muscle cells, in 78% of the epithelial cells, in 78% of the stromal cells and in 67% of the Rokitansky-Aschoff sinuses.
Positive correlations exist between the degree of inflammation in the gallbladder wall and the degree of COX-2 expression in the smooth muscle cells (r= +0.71, p<0.001) and in the vascular smooth muscle cells (r= +0.51, p<0.001).
In 8 gallbladder specimens with gastric metaplasia, an increase of COX-2 expression was seen in 100% of the epithelial cells, in 87% of the smooth muscle cells, in 75% of the vascular smooth muscle cells, in 63% of the stromal cells and in 37% of the Rokitansky-Aschoff sinuses. In this group, a positive correlation was seen between the degree of inflammation in the gallbladder wall and the degree of COX-2 expression in the stromal cells (r= +0.72, p<0.05).
In 13 gallbladder specimens without metaplasia, an increased COX-2 expression was determined in 85% of the smooth muscle cells, in 69% of the epithelial cells, in 69% of the vascular smooth muscle cells, in 54% of the stromal cells and in 38% of the Rokitansky-Aschoff sinuses. In this group, a positive correlation was revealed between the degree of inflammation in the gallbladder wall and the degree of COX-2 expression in the smooth muscle cells (r= +0.82, p<0.05).
In patients with chronic calculous cholecystitis, a negative correlation was revealed between the absorption function of gallbladder and the thickness of the gallbladder wall (r= -0.71, p<0.05) (33).
Obtained data demonstrate:
- The excessive COX-2 expression in the smooth muscle cells, in the vascular smooth muscle cells and in the epithelial cells of the gallbladder may be the cause of chronic aseptic inflammation, of the decrease of water absorption and of the decrease of hepatic bile “passage” into the gallbladder up to 35%.
- The excessive COX-2 expression in the smooth muscle cells may be the cause of gallbladder hypomotility and biliary pain.
- The excessive COX-2 expression in the smooth muscle cells, in the vascular smooth muscle cells and in the epithelial cells of the gallbladder may be the cause of increased thickness of the gallbladder wall.
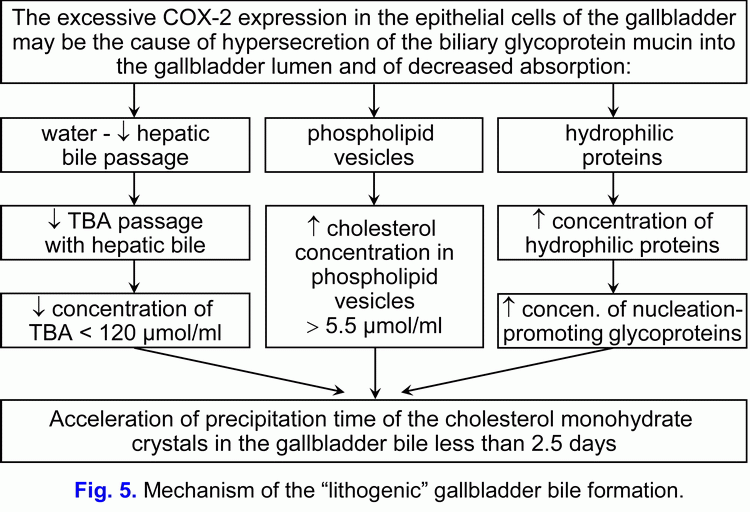
- The excessive COX-2 expression in the epithelial cells of the gallbladder may be the cause of hypersecretion of the biliary glycoprotein mucin into the gallbladder lumen and of increase of the biliary glycoprotein mucin concentration in the gallbladder bile.
Taking account the fact that the excessive COX-2 expression in the smooth muscle cells, in the vascular smooth muscle cells and in the epithelial cells of the gallbladder may appear at the early stage of the development of cholecystolithiasis, then the excessive COX-2 expression in the smooth muscle cells, in the vascular smooth muscle cells and in the epithelial cells of the gallbladder may be the physical cause of appearance of the chronic intrahepatic “bland” cholestasis and the “lithogenic” gallbladder bile formation:
1) to decrease of water absorption by the epithelial cells of mucous gallbladder and to promote the reduce of the hepatic bile inflow into the gallbladder (the limitation of “passive” passage of the hepatic bile) and to promote the reduce of the total bile acids concentration in the gallbladder bile;
2) to decrease of vesicular cholesterol absorption by the epithelial cells of mucous gallbladder and to promote the increase of cholesterol concentration in the phospholipid vesicles in the gallbladder bile;
3) to decrease of hydrophilic proteins absorption by the epithelial cells of mucous gallbladder and to promote the increase of hydrophilic proteins concentration in the gallbladder bile.
This process is accompanied by the increase of the vesicular cholesterol/total bile acids ratio and by the increase of the total biliary proteins/total bile acids ratio and it promote the rise of the rate of cholesterol monohydrate crystals precipitation on the epithelial cells of mucous gallbladder.
Hence, than the less is the absorption of vesicular cholesterol by the epithelial cells of mucous gallbladder, then the higher is the cholesterol concentration in the gallbladder bile and the less is the nucleation time of cholesterol monohydrate crystals in the gallbladder bile, and vice versa. Hence, the excessive COX-2 expression in the epithelial cells of the gallbladder decreases the absorption and concentration functions of the gallbladder and promotes the “lithogenic” gallbladder bile formation. The decrease of the evacuation function of the gallbladder (the excessive COX-2 expression in the smooth muscle cells) is the predisposing factor for the precipitation of cholesterol monohydrate crystals and for the formation of cholesterol gallstones (fig. 5).
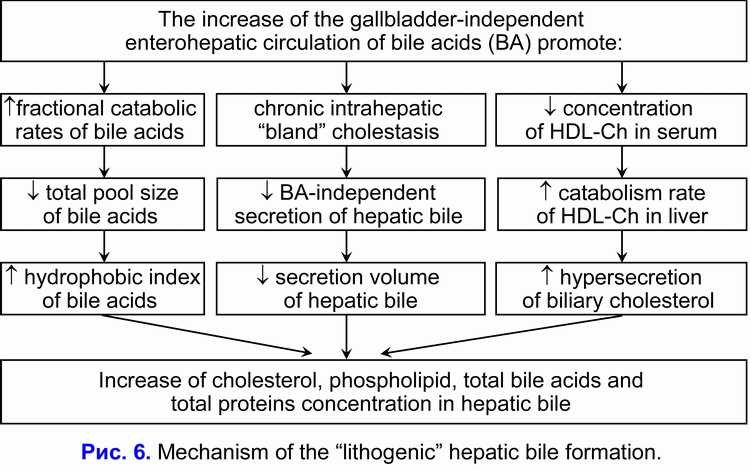
The decrease of hepatic bile “passage” into the gallbladder promote the hepatic bile “passage” into the duodenum and increase the frequency of the gallbladder-independent enterohepatic circulation of bile acids and stimulate the formation of the hydrophobic hepatotoxic deoxycholic bile acid (DCA) (10, 34, 35).
The increase of the frequency of the gallbladder-independent enterohepatic circulation of bile acids and the increase of hydrophobic hepatotoxic deoxycholic bile acid concentration in the hepatocytes reduce the bile-acid-independent secretion of hepatic bile and stimulate the chronic intrahepatic “bland” cholestasis formation (36, 37). Hence, the decrease of hepatic bile “passage” into the gallbladder and, respectively, the increase of hepatic bile “passage” into the duodenum is the cause of the increased frequency of the gallbladder-independent enterohepatic circulation of bile acids and of the appearance of the chronic intrahepatic “bland” cholestasis.
The chronic intrahepatic “bland” cholestasis is accompanied by the reduction of secretion volume of hepatic bile and by the rise of biliary cholesterol, total bile acids and total biliary protein concentration in the hepatic bile (fig. 6) (38, 39). The increase of biliary cholesterol concentration in the hepatic bile promote the rise of the biliary cholesterol concentration in phospholipid vesicles (r= +0.59, p<0.05) (40). The increase of total bile acids concentration in the hepatic bile reduces the stability of phospholipid vesicles and shortens the nucleation time of cholesterol monohydrate crystals (r= -0.53, p<0.05) (40).
We suppose that the chronic intrahepatic “bland” cholestasis, reducing the secretion rate and the hepatic bile volume, promotes the rise of biliary cholesterol, total bile acids and total biliary protein concentration in the hepatic bile, and shortens the nucleation time of cholesterol monohydrate crystals, what predispose to the “lithogenic” hepatic bile formation.
The decrease of absorption, concentration and evacuation functions of the gallbladder promote the “lithogenic” gallbladder bile formation, the chronic intrahepatic “bland” cholestasis promote the “lithogenic” hepatic bile formation (fig. 5, fig. 6). These two factors determine the cholesterol gallstones formation.
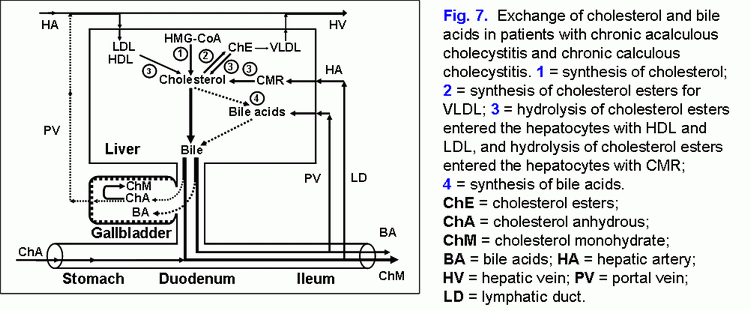
In patients with chronic acalculous cholecystitis with biliary sludge, the formation of cholesterol gallstones is promoted by the decrease of absorption (the decrease of the water and phospholipid vesicles absorption), concentration (the decrease of total bile acids concentration in gallbladder bile) and evacuation (the decrease of the gallbladder-dependent output of biliary cholesterol) functions and by the increase of secretion (hypersecretion of glycoprotein mucin by the gallbladder mucosa) function of the gallbladder (fig. 7) (41).
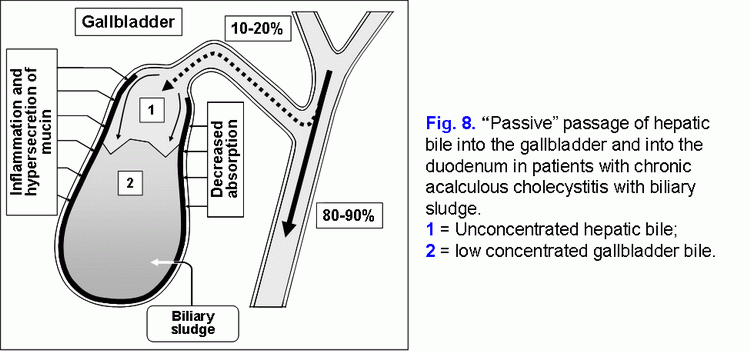
The decrease of the water absorption rate of in the gallbladder wall limits the “passive” passage of the hepatic bile into the gallbladder and increases the hepatic bile passage into the duodenum (fig. 8) (41-43).
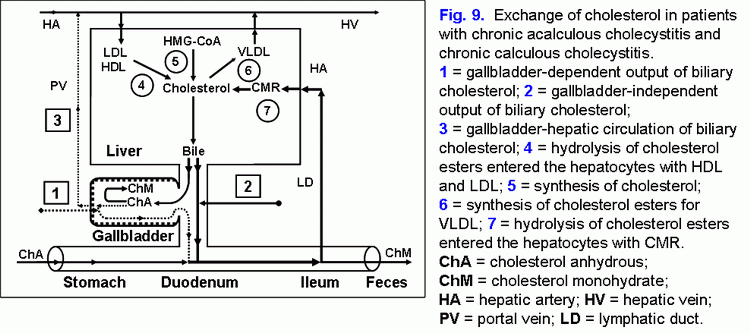
The decrease of the evacuation function of the gallbladder reduces the “active” passage of the hepatic bile into the gallbladder (44, 45). This process is accompanied by the decrease of the total bile acids concentration and the increase of the biliary cholesterol concentration in phospholipid vesicles and it also promotes the increase of time for precipitation of cholesterol monohydrate crystals and the formation of cholesterol gallstones (fig. 9) (46-50).
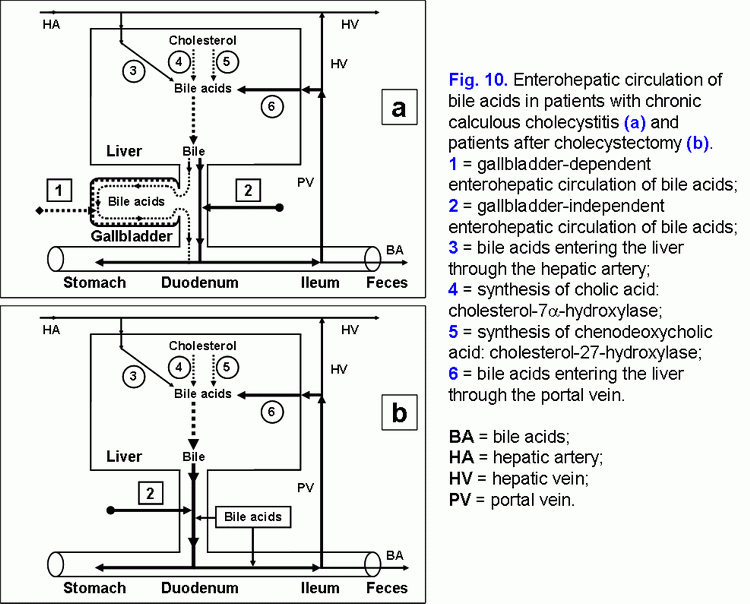
The excessive hepatic bile passage from the liver into the duodenum increases the frequency of the gallbladder independent enterohepatic circulation of bile acids. The gallbladder-independent enterohepatic circulation of bile acids in patients with the cholesterol gallstone disease (CGD) or after cholecystectomy is raised (fig. 10).
It results in: 1) the increase of the hydrophobic hepatotoxic DCA formation (table 3) and its accumulation in hepatocytes (51), 2) the formation of morphological changes in the liver (nonspecific reactive hepatitis) (52) and 3) the appearance of cholestasis (53).
The risk of cancer of the liver, the pancreas, the small intestine, and the colon increases as well (54-62). The increases of DCA, participating in the enterohepatic circulation, and of other toxic agents in the hepatic bile can result in chronic pancreatitis and duodeno-gastral reflux (63-66).
References:
- Hofmann A.F. Bile secretion and the enterohepatic circulation of bile acids. In: Feldman M., Scharschmidt B.F., Sleisenger M.H., eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management. 6th ed. Philadelphia: WB Saunders Company, 1998: 937-948.
- Heuman D.M., Hylemon P.B., Vlahcevic Z.R. Regulation of bile acid synthesis. III. Correlation between biliary bile acid hydrophobicity index and the activities of enzymes regulating cholesterol and bile acid synthesis in the rat. J Lipid Res 1989; 30: 1161-1171.
- Hofmann A.F. Bile Acids. In: Arias I.M., Boyer J.L., Fausto N., Jakoby W.B., Schachter D.A., Shafritz D.A., eds. The Liver, Biology and Pathobiology. 3rd ed. New York: Raven Press, 1994: 677-718.
- Roda A., Piazza F., Baraldini M., Speconi E., Guerra M.C., Cerre C., Forti G.C. Taurohyodeoxycholic acid protects against taurochenodeoxycholic acid-induced cholestasis in the rat. Hepatology 1998; 27: 520-525.
- Scholmerich J., Baumgartner U., Miyai K., Gerok W. Tauroursodeoxycholate prevents taurolithocholate-induced cholestasis and toxicity in rat liver. J Hepatol 1990; 10(3): 280-283.
- Lim A.G., Ahmed H.A., Jazrawi R.P., Levy J.H., Northfield T.C. Effects of bile acids on human hepatic mitochondria. Eur J Gastroenterol Hepatol 1994; 6(12): 1157-1163.
- Paumgartner G., Beuers U. Bile acids and the liver. In: Surrenti C., Casini A., Milani S., Pinzani M., eds. Fat-storing Cells and Liver Fibrosis. Dordrecht: Kluwer Academic Publishers, 1994: 330-339.
- Ochsenkuhn T; Bayerderffer E; Meining A; Schinkel M; Thiede C; Nussler V; Sackmann M; Hatz R; Neubauer A; Paumgartner G. Colonic mucosal proliferation is related to serum deoxycholic acid levels. Cancer 1999; 85(8): 1664-1669.
- Shekels L.L., Beste J.E., Ho S.B. Tauroursodeoxycholic acid protects in vitro models of human colonic cancer cells from cytotoxic effects of hydrophobic bile acids. Lab Clin Med 1996; 127: 57-66.
- Carey M.C., Duane W.C. Enterohepatic circulation. In: Arias I.M., Boyer J.L., Fausto N., Jakoby W.B., Schachter D.A., Shafritz D.A., eds. The Liver, Biology and Pathobiology. 3rd ed. New York: Raven Press, 1994: 719-767.
- Ewerth S., Angelin B., Einarsson K., Nilsell K., Bjorkhem I. Serum concentrations of ursodeoxycholic acid in portal venous and systemic venous blood of fasting humans as determined by isotope dilution-mass spectrometry. Gastroenterology 1985; 88:126-133.
- Malavolti M., Ceryak S., Fromm H. Modulation of bile secretion by hepatic low-density lipoprotein uptake and by chenodeoxycholic acid and ursodeoxycholic acid treatment in the hamster. Gastroenterology 1987; 93: 1104-1115.
- Malavolti M., Fromm H., Ceryak S., Roberts I.M. Modulation of low-density lipoprotein receptor activity by bile acids: differential effects of chenodeoxycholic and ursodeoxycholic acids in hamster. J Lipid Res 1987; 28: 1281-1295.
- Ceryak S., Bouscarel B., Malavolti M., Robins S.J., Fromm H. Effect of ursodeoxycholic bile acid on hepatic LDL metabolism in dietary hypercholesterolemic hamsters. Gastroenterology 1996; 110: A1165.
- Fromm H., Bouscarel B., Ceryak S., Malavolti M. Direct effects of bile acids on low-density lipoprotein metabolism. In: Fromm H., Leuschner U., eds. Bile Acids-Cholestasis-Gallstones. Advances in Basic and Clinical Bile Acid Research. Dordrecht: Kluwer, 1996: 141-144.
- Maton P.N., Ellis H.J., Higgins J.P., Dowling R.H. Hepatic HMG-CoA reductase in human cholelithiasis: effect of chenodeoxycholic and ursodeoxycholic acids. Europ J Clin Invest 1980; 10: 325-332.
- Vlahcevic Z.R., Heuman D.H., Hylemon P.B. Regulation of bile acid synthesis. Hepatology 1991; 13: 590-600.
- Vlahcevic Z.R. Regulation of cholesterol 7a-hydroxylase by different effectors. Ital J Gastroenterol 1996; 28: 337-339.
- Lindbland L., Lundholm K., Schersten T. Influence of cholic and chenodeoxycholic acid on biliary cholesterol secretion in man. Europ J Clin Invest 1977; 7: 383-388.
- Sama C., LaRusso N.F., Loper del Pino V., Thistle J.L. Effects of acute bile acid administration on biliary lipid secretion in healthy volunteers. Gastroenterology 1982; 82: 515-525.
- Carulli N., Loria P., Bertolotti M. Effects of acute change of bile acid pool composition on biliary lipid secretion. J Clin Invest 1984; 74: 614-624.
- Carey M.C. Pathogenesis of gallstones. Amer J Surg 1993; 165: 410-419.
- Carey M.C. Formation and growth of cholesterol gallstones: the new synthesis. In: Fromm H., Leuschner U., eds. Bile Acids-Cholestasis-Gallstones. Advances in Basic and Clinical Bile Acid Research. Dordrecht: Kluwer, 1996: 147-175.
- Lichtenberg D., Ragimova S., Bor A., Almog S., Vinkler C., Peled Y., Halpern Z. Stability of mixed micellar systems made by solubilizing phosphatidylcholine-cholesterol vesicles by bile salts. Hepatology 1990; 12: 149S-154S.
- Van de Heijning B.J.M., Stolk M.F.J., van Erpecum K.J., Renooij W., van Berge Henegouwen G.P. The effects of bile salt hydrophobicity on model bile vesicles morphology. Biochim Biophys Acta 1994; 1212: 203-210.
- Einarsson K., Grundy S.M. Effects of feeding cholic and chenodeoxycholic acid on cholesterol absorption and hepatic secretion of biliary lipids in man. J Lipid Res 1980; 21: 23-34.
- Ponz de Leon M., Carulli N. The influence of bile acid pool composition on the regulation of cholesterol absorption. In: Paumgartner G., Stiehl A., Gerok W., eds. Bile Acids and Lipids. London: MTP Press, 1981: 133-140.
- Sama C., LaRusso N.F. Effect of deoxycholic, chenodeoxycholic, and cholic acids on intestinal absorption in humans. Mayo Clin Proc 1982; 57: 44-50.
- Lanzini A., Northfield T.C. Effect of ursodeoxycholic acid on biliary lipid coupling and on cholesterol absorption during fasting and eating in subjects with cholesterol gallstones. Gastroenterology 1988; 95(2): 408-416.
- Christl S.U., Bartram H.P., Paul A., Kelber E., Scheppach W., Kasper H. Bile acid metabolism by colonic bacteria in continous culture: effects of strach and pH. Ann. Nutrition metabolism 1997; 41: 45-51.
- Farkkila M.A., Turunen V.M., Miettinen T.A. The role of intestinal bacteria in regulation of cholesterol metabolism. Gastroenterology 1994; 106(4): A891.
- Golubev S.S., Kozlova N.M., Turumin J.L., Raevskaja L.Ju. The expression of cyclooxygenase-2 in the gallbladder wall of the patients with chronic calculous cholecystitis. Acta Gastroenterologica Belgica 2007; 70(1): Abstr. 31.
- Kozlova N.M., Turumin J.L., Galeev Y.M., Popov M.V. The functional state of hepato-biliary system in the patients with chronic calculous cholecystitis in the stage of exacerbation. Acta Gastroenterologica Belgica 2007; 70(1): Abstr. 33.
- Roda E. Changes of biliary lipid secretion and bile acid pool size in pathogenesis of cholesterol gallstones. Falk Symposium No. 84: Bile Acids-Cholestasis-Gallstones - Advances in Basic and Clinical Bile Acid Research: Abstr. Book. Berlin, Germany, 1995: Abstr. 26.
- Roda E., Cipolla A., Bazzoli F. et al. Modifications in biliary lipid secretion and bile acid kinetics in the pathogenesis of cholesterol gallstones. In: H. Fromm, U. Leuschner, eds Bile Acids-Cholestasis-Gallstones. Advances in Basic and Clinical Bile Acid Research. Dordrecht: Kluwer, 1996: 176-179.
- Honda A., Yoshida T., Tanaka N. et al. Increased bile acid concentration in liver tissue with cholesterol gallstone disease. J. Gastroenterol. 1995; 30: 61-66.
- Lesage G.D., Schteingart C.D., Hofmann A.F. Effect of bile acid hydrophobicity on biliary transit time and intracellular mobility: a comparison of four fruorescent bile acids analogues. Gastroenterology. 38. 1994; 106(4): Abstr. 929.
- Farrel G.C. Liver disease caused by drugs, anesthetics, and toxins. In: M. Feldman, B.F. Scharschmidt, M.H. Sleisenger, eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management. 6th ed. Philadelphia: WB Saunders Company, 1998: 1221-1252.
- Sherlock S., Dooley J. Diseases of the liver and biliary system. 9th ed. Oxford: Blackweel Scientific Publications, 1993. 649 p.
- Lee S.P., Park H.Z., Madani H., Kaler E.W. Partial characterization of a nonmicellar system of cholesterol solubilization in bile. Amer. J. Physiol. 1987; 252: G374-G384.
- Turumin J.L., Shanturov V.A. Pathogenesis and treatment of cholesterol gallstone disease. XIV International Bile Acid Meeting "Bile Acids in Hepatobiliary Diseases - Basic Research and Clinical Application" (Falk Symposium No. 93). Freiburg, Germany, 1996: Abstr 104.
- Jacyna M.R., Ross P.E., Hopwood H., Bouchier I.A.D. Studies on the mechanism of non-visualization of diseased human gallbladders during oral cholecystography. Postgrad Med J 1988; 64(758): 931-935.
- Turumin J.L., Shanturov V.A. The disturbance of the gallbladder bile formation in-patients with cholesterol gallstone disease. XIV International Bile Acid Meeting "Bile Acids in Hepatobiliary Diseases - Basic Research and Clinical Application" (Falk Symposium No. 93). Freiburg, Germany, 1996: Abstr. 105.
- Jazrawi P.P., Pazzi P., Petroni M.L., Northfield T.C. Postprandial refilling and turnover of bile: a novel approach to assessing gallbladder stasis in cholelithiasis. Gastroenterology 1995; 109: 582-591.
- Carey M.C. Formation and growth of cholesterol gallstones: the new synthesis. In: Fromm H., Leuschner U., eds. Bile Acids-Cholestasis-Gallstones. Advances in Basic and Clinical Bile Acid Research. Dordrecht: Kluwer, 1996: 147-175.
- Carey M.C., Cahalane M.J. Whither biliary sludge? Gastroenterology 1988; 95: 508-523.
- Carey M.C., LaMont J.T. Cholesterol gallstone formation. 1. Physical-chemistry of bile and biliary lipid secretion. Prog Liver Dis 1992; 10: 139-163.
- Carey M.C. Pathogenesis of gallstones. Amer J Surg 1993; 165: 410-419.
- Carey M.C. Pathogenesis of cholesterol and pigment gallstones: some radical new concepts. In: Gerok W., Loginov A.S., Pokrowskij V.I., eds. New Trends in Hepatology 1996. Dordrecht: Kluwer, 1996: 64-83.
- LaMont J.T., Carey M.C. Cholesterol gallstone formation. 2. Pathogenesis and pathomechanics. Progr Liver Dis 1992; 10: 165-191.
- Honda A., Yoshida T., Tanaka N., Matsuzaki Y., He B., Shoda J., Osuga T. Increased bile acid concentration in liver tissue with cholesterol gallstone disease. J Gastroenterol 1995; 30(1): 61-66.
- Geraghty J.M., Goldin R.D. Liver changes associated with cholecystitis. J Clin Pathol 1994; 47(5): 457-60.
- Zubovski G.A., ed. Radio and ultrasonic diagnosis of biliary tract diseases. Moscow: Medicine, 1987: 1-240.
- Bayerderffer E; Mannes GA; Richter WO; Ochsenkuhn T; Wiebecke B; Kepcke W; Paumgartner G. Increased serum deoxycholic acid levels in men with colorectal adenomas. Gastroenterology 1993; 104(1): 145-151.
- Bayerderffer E; Mannes GA; Richter WO; Ochsenkuhn T; Seeholzer G; Kepcke W; Wiebecke B; Paumgartner G. Decreased high-density lipoprotein cholesterol and increased low-density cholesterol levels in patients with colorectal adenomas. Ann Intern Med 1993; 118(7): 481-487.
- Bayerderffer E; Mannes GA; Ochsenkuhn T; Dirschedl P; Paumgartner G. Variation of serum bile acids in patients with colorectal adenomas during a one-year follow-up. Digestion 1994; 55(2): 121-129.
- Bayerderffer E; Mannes GA; Ochsenkuhn T; Dirschedl P; Wiebecke B; Paumgartner G. Unconjugated secondary bile acids in the serum of patients with colorectal adenomas. Gut 1995; 36(2): 268-273.
- Ekbom A., Yuen J., Adami H.-O., McLaughlin J.K., Chow W.-H., Persson I., Fraumeni J.F. Cholecystectomy and colorectal cancer. Gastroenterology 1993; 105: 142-147.
- Goldbohm R.A., van den Brandt P.A., van t Veer P., Dorant E., Sturmans F., Hermus R.J. Cholecystectomy and colorectal cancer: evidence from a cohort study on diet and cancer. Int J Cancer 1993; 53(5): 735-739.
- Johansen C., Chow W.-H., Jorgensen T., Mellemkjaer L., Engholm G., Olsen J.H. Risk of colorectal cancer and other cancers in patients with gallstones. Gut 1996; 39: 439-443.
- Chow W.H., Johansen C., Gridley G., Mellemkjair L., Olsen J.H., Fraumeni J.F. Gallstones, cholecystectomy and risk of cancers of the liver, biliary tract and pancreas. Br J Cancer 1999; 79(3-4) 640-644.
- Strom B.L., Soloway R.D., Rios-Dalenz J., Rodriguez-Martinez H.A., West S.L., Kinman J.L., Crowther R.S., Taylor D., Polansky M., Berlin J.A. Biochemical epidemiology of gallbladder cancer. Hepatology 1996, 23: 1402-1411.
- Barthet M., Affriat C., Bernard J.P., Berthezene P., Dagorn J.C., Sahel J. Is biliary lithiasis associated with pancreatographic changes? Gut 1995, 36(5): 761-765.
- Portincasa P., Di Ciaula A., Palmieri V., Velardi A., VanBerge Henegouwen G.P., Palasciano G. Impaired gallbladder and gastric motility and pathological gastro-oesophageal reflux in gallstone patients. Eur J Clin Invest 1997; 27(8): 653-661.
- Stein H.J., Kauer W.K.H., Feussner H., Siewert J.R. Bile acids as components of the duodenogastric refluate: detection, relationship to bilirubin, mechanism of injury, and clinical relevance. Hepatogastroenterology 1999; 46(25): 66-73.
- Fukumoto Y., Murakami F., Andoh M., Mizumachi S., Okita K. Effects of the elevation of serum bile acids on gastric mucosal damage. Hepatol Res 1999; 14(3): 195-203.
|